一新穎TFG基因突變可造成第二型遺傳性運動感覺神經病變及TFG蛋白功能缺損
A novel TFG mutation causes Charcot-Marie-Tooth disease type 2 and impairs TFG function.
神經內科主治醫師 李宜中
遺傳性運動感覺神經病變 (Hereditary Motor Sensory Neuropathy; HMSN),是一群因不同的致病基因突變所造成的遺傳性周邊神經病變[1]。它最早的完整病例報告描述是在西元1886年由法國的Jean-Martin Charcot和Pierre Marie,以及英國的Howard Henry Tooth所發表,因而它又有另一個更廣為人知的病名,就是由這三位專家的姓氏而來的Charcot-Marie-Tooth disease(CMT)[2]。
CMT的發病年齡從剛出生到老年都有可能,但最常見到的是在兒童、青春期、及年輕成人時期。這類病人的周邊神經會緩慢逐漸地退化,通常由肢體遠端開始,因而所造成的症狀也是緩慢逐漸地發生。常常一開始的症狀是腳掌變形,病人的足弓變得很彎很高,少數人會變成平底足,這是因為腳掌內部肌肉萎縮,因而無法保持正常腳掌的型態。接著小腿的肌肉也會跟著萎縮,下肢肌肉的萎縮常會造成病人行走及平衡的問題,而病人常會跌倒或扭傷足踝。當疾病更為進展時,許多病人需要柺杖或輪椅來幫助行動,同時手掌的肌肉也會萎縮,造成病人寫字等手部精細活動的障礙。由於感覺神經也同時受損,有些病人也會感覺到肢體末端麻木或感覺喪失。感覺的缺失常發生在肢體的遠端,對疼痛、溫度、振動和本體感覺等不同型式感覺常有不同程度的損害。 CMT病人的臨床症狀常發生的非常緩慢,在大部分的病人中,症狀開始的時間常是模糊無法回憶。雖然在所有CMT病人中,詳細的神經學檢查能確定有感覺神經的損害,但只有一小部份病人會有主觀麻木、刺痛、燒灼等不舒服感覺。幾乎所有病人的肌腱反射都下降或消失。有些病人能在皮下觸摸到增大的神經,而顫抖及肌肉痙癴也可在部份病人身上發現[2]。台灣目前並沒有CMT的盛行率資料,但曾在西元1988到1989年間統計520位因多發性神經病變而住院的病人中,22位 (4.23%)是CMT的患者[3]。在西方國家,CMT的盛行率是十萬分之20.1- 40,而CMT1則在每十萬人中有16.2- 30人[4-6]。如果將此數據套用在台灣的人口數上,預計在台灣應有4600至9200 CMT患者。
CMT的分類一直在演變,反映了探索CMT致病基因的相關研究快速地進展。在傳統上,遺傳性運動感覺神經病變可藉由遺傳的型式、發病的年齡、以及電生理和病理的特徵來分類[1-2]。一般可先大略分為脫髓鞘型或軸索病變型,當有了進一步詳細家族史和發病年齡資料就可進一步分類。第一型遺傳性運動感覺神經病變 (CMT1)是在兒童期或之後發病的體顯性遺傳的脫髓鞘型神經病變,而第二型遺傳性運動感覺神經病變 (CMT2)是體顯性或體隱性遺傳的軸索病變型神經病變。在臨床診斷病人時,神經傳導測試是區分脫髓鞘型或軸索病變型CMT最為方便有效率的方法。目前學界所公認用來區分脫髓鞘型或是軸索病變型CMT的電生理參數是正中神經運動神經傳導速度,正中神經運動神經傳導速度在脫髓鞘型CMT的病人中常是低於38 m/s,而在軸索病變型CMT的病人中常是高於38 m/s[7]。現今CMT的分類趨勢是在傳統的分類下再根據不同的致病基因或其基因位置(Locus)來做進一步的分類。如在CMT1中,因PMP22基因重複所致病者分類為CMT1A,因MPZ基因突變所致病者分類為CMT1B,而因MFN2基因突變所導致的CMT2則分類為CMT2A。目前已有超過80種不同CMT的致病基因已被發現[8-9]。
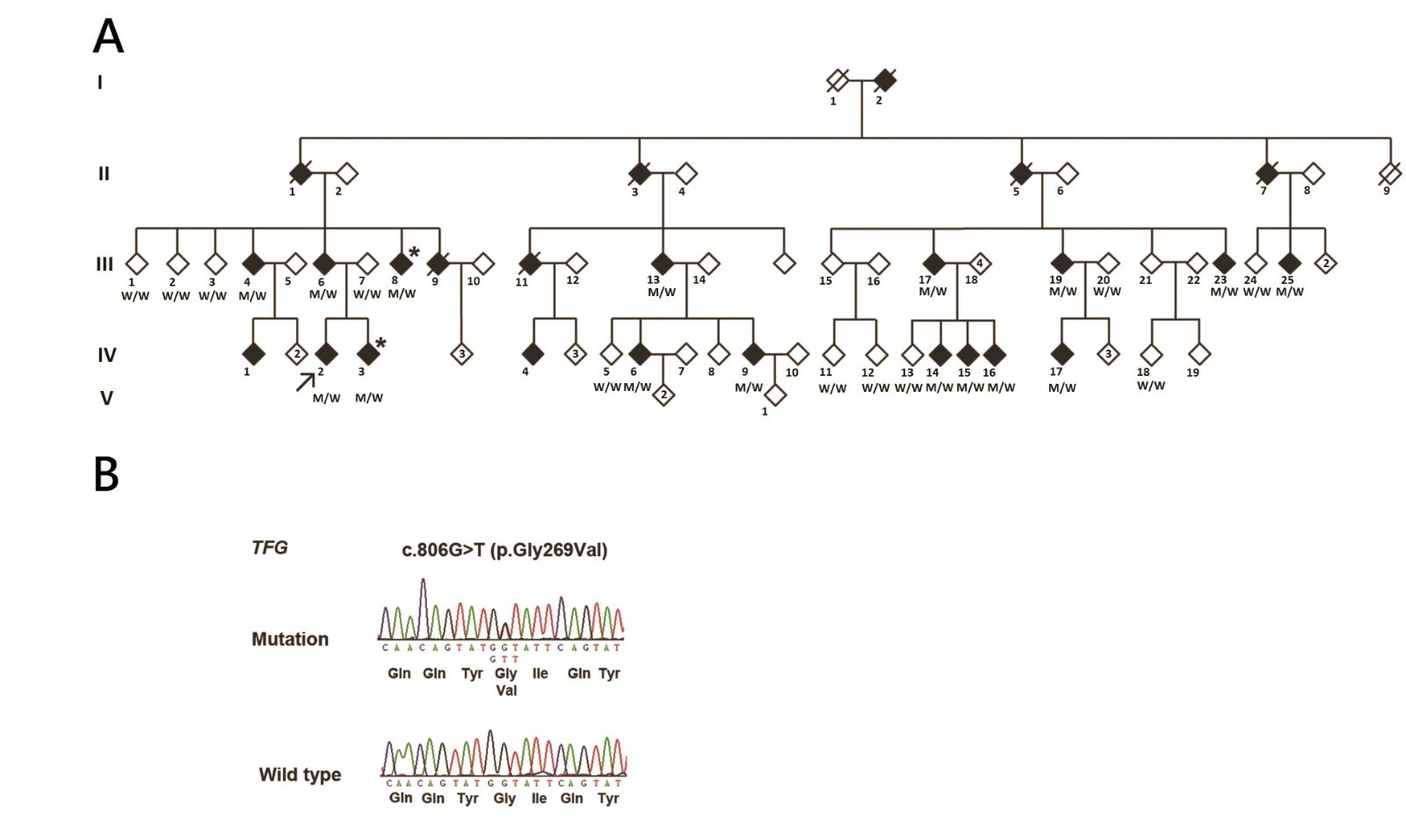
在世界各族群的CMT病人中,各CMT致病基因發生突變的比例雖有些微不同,最常見的突變基因都是下列4種:PMP22 (CMT1A)、GJB1(CMTX1)、MPZ (CMT1B)、以及MFN2 (CMT2A)。通常這些基因突變的病例數佔據所有能有基因診斷的病例數的八、九成以上。由英美兩大研究團體發表了他們對於CMT病人進行廣泛深入的各類致病基因篩檢的結果來看,其中大部分致病基因突變所佔的比例都小於百分之一,而仍有三分之一以上的病人找不出致病突變 (表一)。我們從十年前就開始募集台灣本土的CMT病例,而從2005年起就開始對大部分所有已知可能造成顯性遺傳CMT的致病基因以及GJB1基因在CMT病人中逐一檢測。目前,我們已在300位彼此無親屬關連的CMT病人中篩檢了20種可能的致病基因,包括PMP22(duplication 和point mutation)、MPZ、CX32、EGR2、NEFL、LITAF、FBLN5、GDAP1、MFN2、KIF1B、RAB7、TRPV4、GARS、NEFL、HSPB1、HSPB8、AARS、DNM2、YARS、以及 KARS等基因[10]。雖然我們花了很大的功夫,仍有99位病人(33%)無法找出他們的致病突變。 而其中有一個CMT2的病人家系特別大(圖一,A)。在此家系中,病人的發病年齡從28歲起到40歲,正中神經運動神經傳導速度的範圍從43.6到60.8 m/s。在此家系中,我們共招募了27位家族成員,包括16位病人及11位未帶病者。而後,我們運用次世代核酸定序 (Next Generation Sequencing; NGS) 技術進行其中兩位病人的全外顯子定序 (Exome sequencing)以尋找此家系內CMT2的致病基因。
在此我先介紹一下什麼是Exome sequencing? Exon是genomic DNA中個別會轉錄成mRNA的部分,而所有exon的集合就稱為exome。所以exome包含了genomic DNA中所有能解碼為蛋白質胺基酸序列的訊息。 Exome sequencing是結合應用whole exome capture及次世代核酸定序技術,先將genomic DNA樣本中exomic DNA先捕捉放大後予以完整定序。捕捉的方法是將genomic DNA碎裂成片段而後與預先製備好的exome–specific probes (針對約165000 exon regions,這可以是包含在溶液中或是chip上)雜合後純化放大。 放大後的exomic DNA經NGS就可以得到Exome詳細的核苷酸序列了[11-12]。
運用Exome sequencing技術找出遺傳疾病之未知致病基因的理想條件是研究對象是一個具有數位病人的家族,如同我們這個的CMT2家系。我們首先挑出兩位親緣關係較遠的病人進行Exome sequencing,這是因為這兩位病人應該具有相同的致病突變,而我們希望能在較少數目的兩病人共同所有的基因變異中分析挑出致病突變。接著,我們將這兩位的exome序列與人類標準genome序列進行比對後,將兩位病人與人類標準序列相異的序列位點變異(variants)彼此比對,所得到的相同variants內應包含致病突變。我們可以進一步利用記載人群內SNP及exome序列的資料庫,如dbSNP、the 1000 Genomes Project、及the Exome Variant Server of the NHLBI Exome Sequencing等,排除已知不會造成所研究疾病的variants,進而窄化致病突變的可能範圍。由於我們所研究的CMT2是體顯性遺傳的疾病,其致病突變應為heterozygous,所以我們只考慮heterozygous mutation。進一步,我們將可能是致病突變的核苷酸變異在所研究的其他家族成員中檢視,致病突變應該與遺傳疾病以符合遺傳模式的方式共同出現在此家系中。最後,我們發現僅有一種核苷酸變異符合所有上述條件,它就是此CMT2的致病突變,在TFG基因上的c.806G>T,p.G269V突變(圖一B)。分析流程及相關數據請見表二。
為了進一步証明TFG基因突變是造成此家系CMT的致病基因,我們運用HumanCytoSNP-12 BeadChip (Illumina, San Diego, CA)對此家系成員身上約30萬個單核苷酸多型性(SNP)的基因標誌進行測定,而後運用這些訊息進行此CMT2家系的遺傳連鎖分析,發現僅有一個染色體區域的LOD score大於3,表示致病基因位於此區域內。此區域為3號染色體108.57cM-114.23cM,而TFG就位於此區域內。
在證實TFGp.G269V突變是造成CMT2的新穎病因後,我們對於此突變的致病機制深感興趣。正常的TFG蛋白會在細胞內的內質網的蛋白質分泌出口上形成八元體 (octamer),調控內質網內所生成蛋白質的分泌。我們首先在HEK293細胞內表現突變的G269V TFG蛋白,而發現此突變蛋白容易在細胞內聚集形成內涵體而使細胞質內具正常功用的可溶型式TFG蛋白量減少。當在細胞內同時表現正常及G269V TFG蛋白時,突變蛋白會與正常TFG蛋白一起形成內涵體而大量減少可溶型式的正常TFG蛋白量。當運用siRNA降低細胞內生性TFG蛋白表現時,細胞的存活下降且蛋白的分泌功能降低。這些缺損可藉由補充表現正常的TFG蛋白而矯正,但補充表現突變的G269V TFG蛋白無助於矯正這些缺損。
根據以上的發現,我們推論我們所發現的TFG p.G269V突變,會影響TFG蛋白的立體構型進而使其容易發生聚集而形成大量內涵體,而使真正能發揮生理功能的TFG蛋白分子數量不足,進而導致其相關的細胞功能障礙。由於具有長軸索的周邊神經對蛋白質分泌功能的依賴性最大,它的突變就以軸索病變型周邊神經病變來表現。我們這個研究論文發表在2014年8月的神經學雜誌 [13]。
我們這個研究能夠完成要感謝病人的配合,我的兩位Mentors宋秉文教授及林恭平醫師多年來對我分別在臨床基因醫學及周邊神經疾病領域的傾囊相授,陽明大學黃彥華博士及劉孜孜博士所進行的exome sequencing的執行與分析,陽明大學范明基老師及國衛院林秀芳老師的寶貴意見,以及我們神經基因實驗室全體成員的努力。希望在未來,我們在這主題上能有更一步的研究與發現。
參考資料
1. Berciano J, Combarros O. Hereditary neuropathies. Curr Opin Neurol 2003; 16:613-22.
2. Shy ME, Lupski JR, Chance P, et al. Hereditary motor and sensory neuropathies: an overview ofclinical, genetic, electrophysiologic, and pathologic features. In: Dyck PJ, Thomas PK, eds. Peripheral Neuropathy, 4rd ed. Philadelphia: Elsevier Saunders, 2005; 1094-136.
3. Lin KP, Kwan SY, Chen SY, et al. Generalized neuropathy in Taiwan: an etiologic survey. Neuroepidemiology 1993;12:257-61.
4. Brust JC, Lovelace RE, Devi S. Clinical and electrodiagnostic features of Charcot-Marie-Tooth syndrome. Acta Neurol Scand 1978; Suppl 68:1-142.
5. Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth disease. Clin Genet 1974; 6:98-118.
6. Holmberg BH. Charcot-Marie-Tooth disease in northern Sweden: an epidemiological and clinical study. Acta Neurol Scand 1993;87:416-22.
7. Shy ME, Garbern JY, Kamholz J. Hereditary motor and sensory neuropathies: a biological perspective. Lancet Neurol 2002; 1:110-18.
8. Murphy SM, Laura M, Fawcett K, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 2012;83:706-10.
9. Saporta AS, Sottile SL, Miller LJ, et al. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 2011;69:22-33.
10. Lin KP, Soong BW, Yang CC, et al. The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PLoS One 2011;6:e29393.
11. Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet 2010;19:R145-51.
12. Kuhlenbäumer G, Hullmann J, Appenzellerm S. Novel genomic techniques open new avenues in the analysis of monogenic disorders. Hum Mutat 2011;32:144-51.
13. Tsai PC, Huang YH, Guo YC, Wu HT, Lin KP, Tsai YS, Liao YC, Liu YT, Liu TT, Kao LS, Yet SF, Fann MJ, Soong BW*, Lee YC*. A novel TFG mutation causes Charcot-Marie-Tooth disease type 2 and impairs TFG function. Neurology 2014;83:903-12.
表一,在我們族群及美英CMT病人中各亞型及致病基因發生突變的比例
|
Subtype or causative gene |
USA |
UK |
Our Group |
|
CMT1A (PMP22 duplication) |
290 (36.9%) |
168 (39.5%) |
132 (44%) |
|
CMT1B (MPZ) |
45 (5.7%) |
6 (1.4%) |
11 (3.7%) |
|
CMT1C (LITAF) |
5 (0.6%) |
4 (0.9%) |
1 (0.3%) |
|
CMT1D (EGR2) |
1 (0.1%) |
0 |
2 (0.7%) |
|
CMT1E (PMP22 point mutation) |
5 (0.6%) |
6 (1.4%) |
4 (1.3%) |
|
CMT1F (NEFL) |
1 (0.3%) |
||
|
CMT1 (FBLN5) |
2 (0.7%) |
||
|
CMTX (GJB1) |
80 (10.2%) |
46 (10.8%) |
27 (9%) |
|
CMT2A (MFN2) |
21 (2.7%) |
12 (2.8%) |
8 (2.7%) |
|
CMT2C (TRPV4) |
3 (0.7%) |
0 |
|
|
CMT2D (GARS) |
3 (0.4%) |
0 |
|
|
CMT2E (NEFL) |
4 (0.5%) |
2 (0.5%) |
7 (2.3%) |
|
CMT2F (HSPB1) |
2 (0.5%) |
1 (0.3%) |
|
|
CMT2I (MPZ) |
1 (0.3%) |
||
|
CMT2K (GDAP1) |
5 (0.6%) |
2 (0.5%) |
1 (0.3%) |
|
CMT2N (AARS) |
1 (0.3%) |
||
|
DI-CMT (GNB4) |
2 (0.7%) |
||
|
RAB7, KIFIB, HSPB8, DNM2, YARS, KARS, MTMR2, SH3TC2, PRX, FIG4 |
7 (0.9%) |
6 (1.4%) |
0 |
|
Unknown |
260 (33%) |
159 (37.4%) |
99 (33%) |
|
Total |
787 |
425 |
300 |
ANN NEUROL 2011;69:22-33; JNNP 2012;83:706-10
表二,第二型遺傳性運動感覺神經病變家系中兩位病人的exome sequencing分析流程及結果
|
|
IV-1 |
IV-4 |
|
Total bases sequenced |
30,658,115.6 Kb |
30,862,079.4 Kb |
|
Raw heterozygous variants (number) |
15,869 |
15,238 |
|
Common heterozygous variants |
6,388 |
|
|
Variants not in dbSNP or 1000 genomes database |
163 |
|
|
Variants not found in other 24 non-CMT patients’ exome databases |
97 |
|
|
Variants resulting in amino acid sequence changes |
57 |
|
|
Variants completely segregating with CMT phenotype in the pedigree |
1 |
|
圖一,第二型遺傳性運動感覺神經病變家系之家系圖 (A) 及突變點相關核苷酸序列 (B)
最後更新: