法布瑞氏症 Fabry Disease
病因:
法布瑞氏症是一種罕見的遺傳性溶小體儲積症(lysosomal storage disorders, LSDs)。罹患此病的病患是因負責製造α半乳糖肝酵素(α-galactosidase A ,簡稱α-GAL)的基因缺陷引起。由於缺乏這種酵素,使得一些醣脂質,特別是globotriaosylceramide (簡稱GL3或Gb-3)無法被代謝,進而堆積在細胞內的溶小體(lysosome)內。而溶小體在細胞中功能有如「回收中心」。溶小體內含有多種酵素可以將大分子物質如蛋白質等消化成小分子,以便細胞再利用。而法布瑞氏症由於基因變異導致無法代謝Gb-3,使得Gb-3堆積在全身許多細胞內的溶小體內,進而造成全身性器官病變。當無法分解的Gb-3不停在細胞內推堆積則會引起發炎反應及破壞,疾病初期還是可逆性的變化,一旦持續被破壞的細胞組織進展成無法修復而結疤、纖維化就會發展成不可逆且無法恢復的傷害。這也是本疾病對腎臟、心臟與腦血管造成併發症的主要原因。其堆積物也會造成周邊神經病變,造成手腳疼痛的症狀。
在台灣的法布瑞氏症確診者,約八成都是非典型法布瑞氏症(IVS4+919G>A),缺乏典型的法布瑞氏症其他的臨床症狀,病患臨床表徵多為心臟症狀為主,又稱為心臟型法布瑞氏症(IVS4)。發病年齡多為40歲以上成年人,且會隨著年紀增長,症狀嚴重程度迅速上升,需提早檢查並定期追蹤,千萬不要輕忽!
法布瑞氏症屬於性聯遺傳疾病,缺陷基因位於X性染色體上,因此對男性的影響會較大(因為男性只有一個 X 染色體,女性則有二個 X 染色體)。在美國約有四萬分之一的男性罹患本病,而且是不分種族的。女性帶原者(一個X 染色體缺陷,而另一個則正常)有時可出現疾病的某些症狀,但通常較輕微。其遺傳模式為若父親X 性染色體帶有法布瑞氏症基因,則只會遺傳給女兒;若母親其中一個X 性染色體帶有法布瑞氏症基因,則有50%機率遺傳給下一代的兒女們。
目前治療可分為「症狀治療」及「酵素替代療法(Enzyme Replacement Therapy)」:
酵素替代療法:酵素替代療法為健保給付藥物,但病患須作藥物審查申請,依照現行健保法規規範病患須符合相對應臨床症狀(如心室肥大,蛋白尿等等),並依照病患基因型態做病理切片(如:心臟或腎臟切片),資料收集完整後才可向健保署申請酵素藥物治療,目前已有許多患者使用酵素替代療法使得病情控制穩定。
症狀治療:神經痛可透過各種藥物來緩解,心臟預防性用藥、腎衰竭要靠血液透析等。
以下為常見的問題~ Q and A ~
Q: 法布瑞氏症是甚麼?為甚麼會發生?
A:
法布瑞氏症為一種罕見之遺傳性疾病,因製造α-半乳糖苷酵素(α-Galactosidase)的基因缺陷, 而導致體內一些醣脂質,特別是 globotriaosylceramide (Gb-3)無法被代謝,因而堆積在全身許多細胞內的溶小體內, 進而造成全身性器官病變。
Q: 典型的法布瑞氏症會出現哪些症狀?
A: 症狀機轉請見以下圖表說明
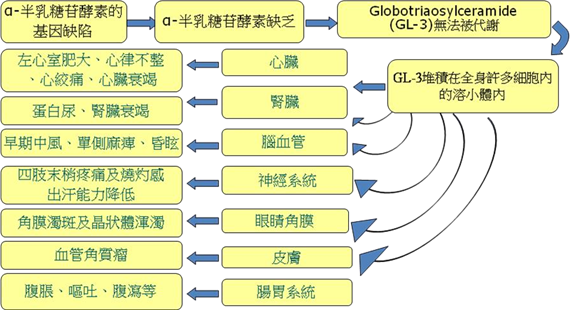
典型的臨床症狀會在兒童或青少年期腳部或手部出現間歇性的疼痛或感覺異常,病人有時會描述像燒灼的感覺,而這種痛有時痛到相當厲害,讓病人無法進行手邊的工作。其疼痛持續的時間從數分鐘到數天都有,有時會重覆性的出現。患者通常在溫度較高或季節變化時較易出現疼痛,也有許多人在運動或上完體育課後會更痛。這種疼痛常被誤診為風濕病、幼年性關節炎、關節痛、生長痛或是心因性疼痛,甚至被認為是裝病或小孩不肯上學所編出來的藉口。此病通常直到成人後才被診斷出來。
排汗能力異常經常出現在兒童期,部分患者(多為典型基因患者)在幼年時期就幾乎不排汗,因此可能容易引起發燒,以及較無法忍受高溫環境及需要消耗體能性的活動,而發燒又會誘發疼痛出現。
另外一項病徵為在下腹、大腿、陰囊、外生殖器的皮膚上出現紫黑色的皮膚病變,稱為血管角質瘤(angiokeratoma)。皮膚病變通常隨年齡增大而增加,但也不一定出現。耳朵、口腔黏膜、結膜、手、指甲也會出現病變。
針對部分患者(多為典型基因病患)眼睛有可能會出現漩渦狀角膜病變(corneal whorls)、窩狀角膜濁斑(cornea verticillata)及視網膜內血管異常彎曲(tortuous retinal blood vessels)等症狀,不過目前研究以上眼睛症狀對於視力影響尚無證據顯示,多屬於良性病徵。耳朵症狀可能會出現耳鳴、聽力受損或突發性耳聾等症狀。
Gb-3 會堆積在組織中並造成血管阻塞。在中樞神經系統會造成中風(比正常人高出10倍),在心臟會造成胸悶、胸痛、心律不整、心室肥大、心室中膈增厚、心肌纖維化或缺氧等,在腎臟會造成蛋白尿、腎功能退化,甚至需要洗腎。通常直到成年(約 20 - 30 歲)才會出現這些症狀,不過 Gb-3 的堆積卻是在出生或更早就開始進行。病患甚至早在 16 歲就會出現腎衰竭,顯示本病病程的變化無常。
臨床診斷要以家族病史、四肢疼痛、皮膚病變、特有的渦狀角膜濁斑,及在尿道沉渣或組織檢體中發現充滿脂質的細胞為基礎。再以生化反應分析法來測量α-GAL酵素的含量及基因檢查作最後確診。
Q: 怎麼樣才能確定診斷是法布瑞氏症?
A:
初步診斷
- 眼睛檢查 : 角膜、結膜、視網膜、晶狀體檢查
- 腎功能檢查 : 尿液蛋白質、肌酸酐清除率、血液尿素氮與肌酐酸檢查
- 心臟與腦血管功能檢查 : 胸部 x 光、心臟超音波、心電圖、核磁共振
- 病理檢查 : 心臟/腎臟切片檢體
確認檢查
- 酵素活性檢測
α-半乳糖苷酵素可藉由血漿、血清、白血球或皮膚纖維母細胞,培養檢測活性。男性患者的酵素活性<1%;心臟變異型患者的酵素活性<10%。女性帶因者的酵素活性差異大,與女性 x 染色體的隨機不活化有關(女性帶因者無法藉此完全確認出來)
基因分析
法布瑞氏症的基因位於 X 染色體的長臂上,稱為 GLA 基因,至今已超過 300 多各突變點被確認出來。台灣常見基因突變分類有:典型、腎臟型、心臟型(IVS4)及腦中風型等。
目前臨床確診大多數是由新生兒篩檢的自選項目「溶小體儲積症(LSD) 」,再經由新生兒轉介單返診做基因檢查後才發現家族中有帶法布瑞氏症基因缺陷,也有少數病患是在門診因臨床症狀符合又做了許多檢查及基因檢驗後才確診。
Q: 法布瑞氏症會遺傳嗎?
A: 本病的基因位於 X 染色體上,因此對男性的影響會較大(因為男性只有一個 X 染色體,女性則有二個 X 染色體);女性帶原者(一個 X 染色體缺陷,而另一個則正常),有時可出現疾病的某些症狀,但通常較輕微。
Q: 法布瑞氏症目前的治療方式?
A: 治療方式包括:
- 症狀治療 : 聽力損傷用助聽器治療,神經痛可透過各種藥物來緩解,腎衰竭靠使用血液透析,心臟問題可能需要心律調節器。
- 酵素替代療法 : 現有二種酵素製劑已獲衛福部核准治療法布瑞氏症之罕見疾病用藥
Q: 怎麼樣監測法布瑞氏症的治療是有效的?
A: 建議每 3-6 個月監測:
腎功能檢查:尿液蛋白質、腎絲球過濾率(GFR)、血液尿素氮與肌酐酸檢查
心臟檢查 :胸部X光、心臟超音波、心電圖、心臟核磁共振等
監測臨床症狀改善程度:眼睛、皮膚、神經疼痛症狀(疼痛評估指標)、生活滿意度(Quality Of Life)等
副作用監測 (如:IgG 抗體、其他副作用症狀等)
血液及尿液中 Gb-3 濃度
Q: 飲食需注意什麼?
A: 飲食控制有助於減輕腎臟(血液透析)的負擔,及改善健康狀態。
原則上需限制水分與含高鉀、高磷、高鈉的食物,蛋白質以動物性蛋白質為主,並且需要攝取足夠的熱量。
Q: 法布瑞氏症能治癒嗎?
A: 目前尚無法徹底治癒
患者需終身使用藥物控制病情,並且越早治療,預後將會越好。
若有男性或女性出現不明原因的四肢疼痛、皮膚病變、排汗能力減少、特有渦狀角膜濁斑、早期中風、左心室肥大、或腎臟功能不全等症狀,應懷疑罹患法布瑞氏症
唯有早期發現,早期施以酵素治療,並耐心治療,與醫師有良好的互動配合,才能達到疾病的控制與生活品質的改善。
國內外相關資源網站
國內-
*台北榮民總醫院遺傳諮詢中心/兒童醫學部
http://wd.vghtpe.gov.tw/gcc/
http://wd.vghtpe.gov.tw/ped/
Tel:02-2871-2121 #1020
*中華民國先天及代謝疾病關懷之友協會
http://www.pku.org.tw/
Tel: 02-6611-5889
*台灣法布瑞氏症病友協會 Taiwan Association of Fabry Disease
http://www.tafd.org.tw/ap/index.aspx
Tel: 02-2383-1330
國外-
*美國法布瑞氏症病友組織 Fabry support & information group
www.fabry.org
~台北榮民總醫院 遺傳諮詢中心關心您~
最後更新: